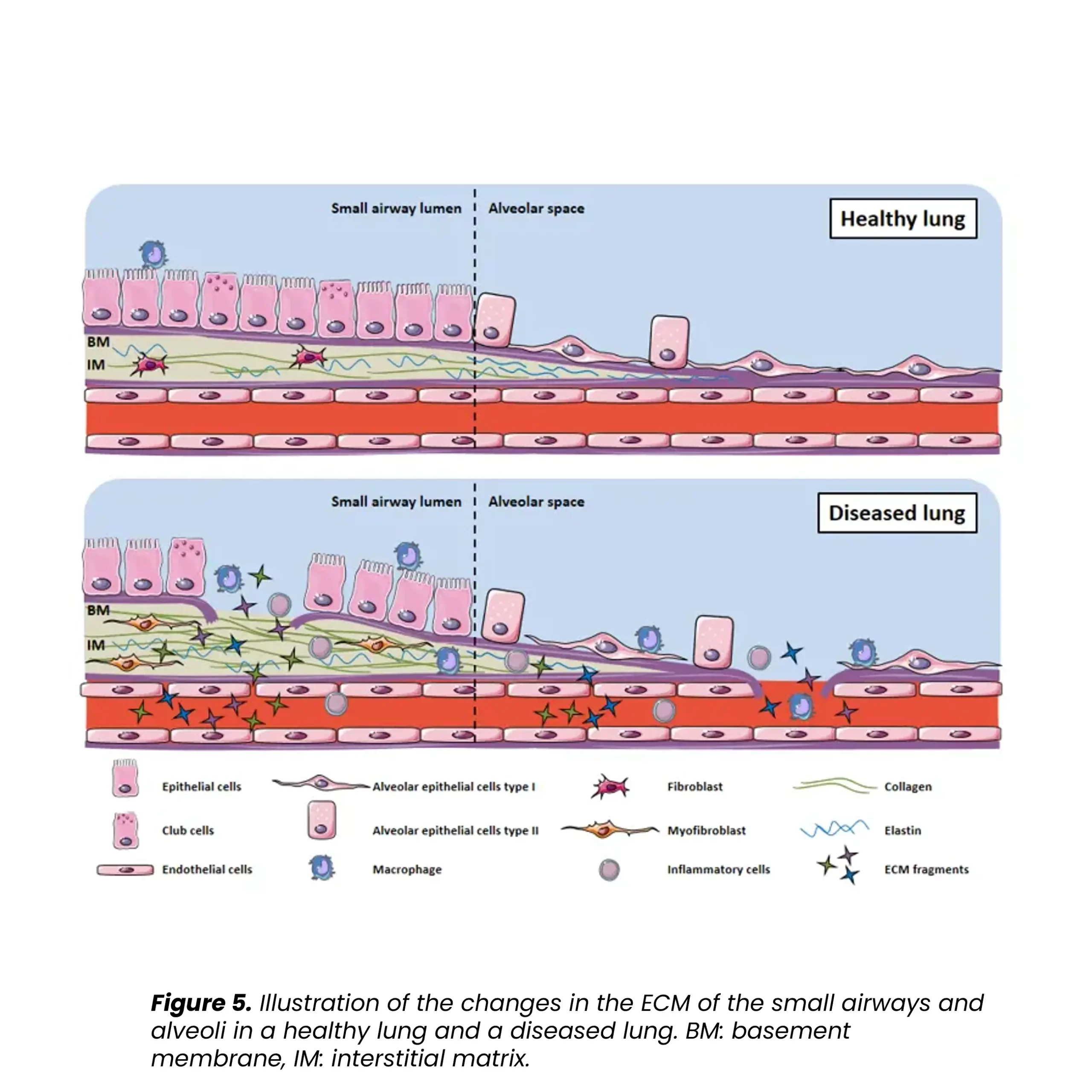
Progressive pulmonary fibrosis, including idiopathic pulmonary fibrosis (IPF), is associated with abnormal lung tissue remodeling and excessive extracellular matrix deposition. The Nordic ProteinFingerPrint TechnologyTM offers extracellular matrix (ECM) biomarkers that quantify the synthesis and degradation of key proteins in the fibrotic lung.
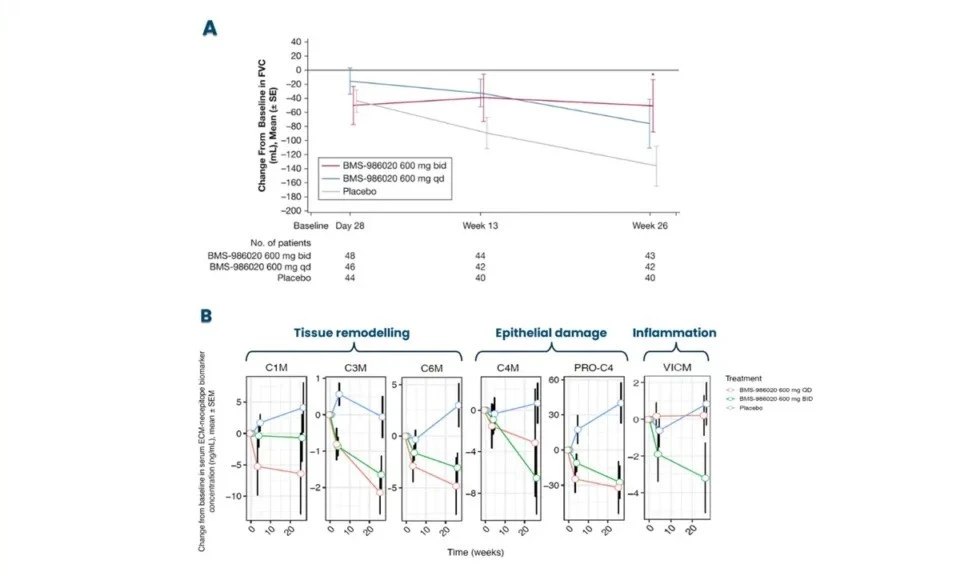
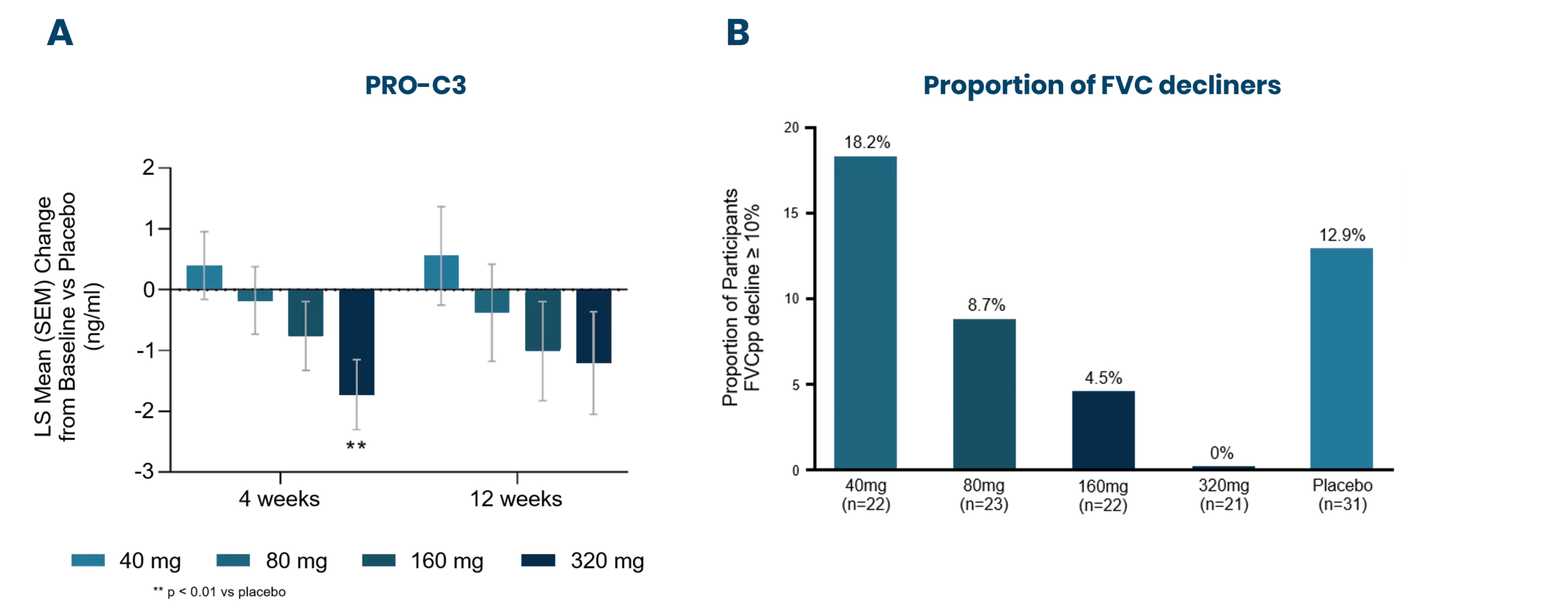
These biomarkers can be used early on for patient stratification, aiding in the inclusion of fast-progressing patients in clinical trials. The most common primary endpoint in clinical trials for IPF is to slow the decline in lung function (forced vital capacity [FVC]).
Nordic Bioscience’s biomarkers have the potential to reflect treatment effects faster and more precisely than spirometry, enabling shorter and more effective trials. These biomarkers can also be used for precision medicine by monitoring and continuous evaluation of anti-fibrotic effects and potentially aid in therapy selection for the individual patient. Additionally, the biomarkers are translational and can be used across preclinical model systems to demonstrate anti-fibrotic effects throughout the drug development pipeline.